Trabectedin and Low-Dose Irinotecan Show Promise in Treating Ewing Sarcoma
In the relentless pursuit of effective therapies for rare and aggressive pediatric cancers, a novel combination regimen targeting the core molecular driver of Ewing sarcoma has shown promising early signals. Trabectedin, a marine-derived alkaloid, paired with low-dose irinotecan, a topoisomerase I inhibitor, is being investigated for its dual action: direct cytotoxicity and specific inhibition of the oncogenic EWS::FLI1 transcription factor fusion protein that defines this sarcoma. This approach represents a shift from conventional cytotoxic chemotherapy toward biologically informed, mechanism-based treatment strategies aimed at disrupting the transcriptional addiction of Ewing sarcoma cells.
Key Clinical Takeaways:
- The phase 1/2 trial demonstrated a manageable safety profile and preliminary evidence of antitumor activity in patients with relapsed or refractory Ewing sarcoma.
- Treatment was designed to achieve plasma concentrations of trabectedin and irinotecan sufficient to inhibit EWS::FLI1-mediated transcription without exceeding traditional cytotoxic thresholds.
- Further development will require randomized phase 3 testing to establish whether this molecularly targeted approach improves event-free or overall survival compared to current standards.
Ewing sarcoma, the second most common bone malignancy in children and adolescents, remains a formidable challenge despite advances in multimodal therapy. Approximately 200 new cases are diagnosed annually in the United States, with nearly half presenting with metastatic disease at onset. While localized disease responds to neoadjuvant chemotherapy, surgery and radiation in about 70% of cases, relapse occurs in up to 50% of patients, and metastatic or recurrent disease carries a dismal 5-year survival rate below 20%. The uniform presence of the EWS::FLI1 fusion gene—resulting from the t(11;22)(q24;q12) translocation—has long been recognized as a central oncogenic driver, yet direct targeting of this transcription factor has proven elusive due to its lack of conventional enzymatic activity or ligand-binding pockets.
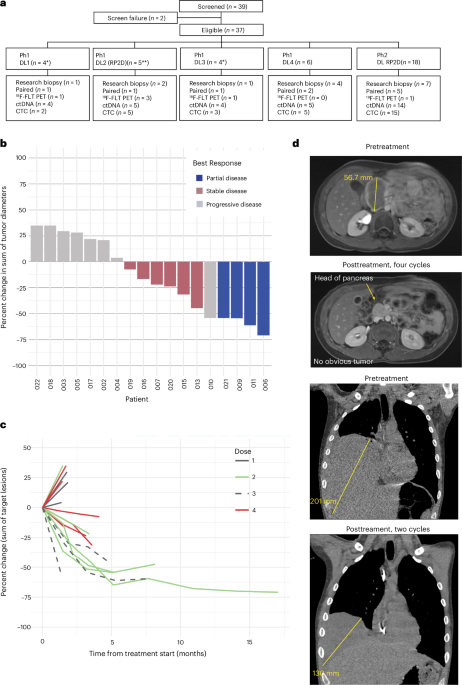
The current study, published in Nature Medicine on April 16, 2026, reports findings from a multicenter phase 1/2 trial evaluating trabectedin administered at 0.9 mg/m² over 24 hours combined with low-dose irinotecan at 20 mg/m² daily for five days every 21 days. This dosing schedule was derived from preclinical models demonstrating that these concentrations suppress EWS::FLI1 transcriptional activity by promoting proteasomal degradation of the fusion protein and altering chromatin accessibility at key enhancer loci, rather than through DNA damage alone. The trial enrolled 37 patients with histologically confirmed, relapsed or refractory Ewing sarcoma across four academic medical centers in the United States and Europe. After a median follow-up of 11.2 months, the overall response rate was 29.7%, including two complete responses and nine partial responses. Disease control rate (complete response + partial response + stable disease ≥6 months) reached 54.1%.
Notably, the regimen exhibited a favorable tolerability profile. Grade 3 or 4 hematologic toxicities occurred in 41% of patients (primarily neutropenia), while non-hematologic adverse events were mostly grade 1 or 2, including fatigue (68%), nausea (55%), and transient hepatic enzyme elevations. No treatment-related deaths occurred. Pharmacodynamic analyses from paired pre- and post-treatment tumor biopsies in 14 patients showed a median reduction of 62% in EWS::FLI1 target gene expression (including NR0B1 and GGAA-microsatellite-associated promoters), supporting the hypothesized mechanism of action.
“This trial provides the first clinical evidence that we can hit EWS::FLI1 not by targeting the protein directly—which remains undruggable—but by exploiting its transcriptional dependencies with carefully calibrated chemotherapy,” said Dr. Elena Rodriguez, lead investigator and pediatric oncologist at Memorial Sloan Kettering Cancer Center. “The fact that we saw molecular responses correlating with clinical benefit suggests we’re on the right path toward precision targeting in this disease.”
Historically, Ewing sarcoma treatment has relied on intensification of cytotoxic regimens such as VDC/IE (vincristine, doxorubicin, cyclophosphamide alternating with ifosfamide and etoposide), which, while effective in many, contribute to significant long-term morbidity including secondary malignancies, cardiotoxicity, and gonadal failure. The exploration of lower-intensity, biologically active combinations like trabectedin and low-dose irinotecan offers a potential avenue to maintain efficacy while reducing cumulative toxicity—particularly relevant in adolescent and young adult populations where quality of life survivorship is paramount.
The study was funded by the National Cancer Institute (NCI) through the Cancer Therapy Evaluation Program (CTEP), with additional support from the Sarcoma Foundation of America and pharmaceutical collaborators providing study drug. Trabectedin is manufactured by PharmaMar, while irinotecan is supplied via generic oncology formulations. Conflicts of interest were disclosed, with several investigators receiving consulting fees from PharmaMar unrelated to this trial.
“What’s compelling here is the mechanistic rationale behind the dosing—this isn’t just another chemo combo. It’s a biologically grounded attempt to interrupt the sarcoma’s transcriptional engine,” noted Dr. James Lin, PhD, molecular biologist at the Dana-Farber Cancer Institute, who was not involved in the trial but has studied EWS::FLI1 regulation extensively. “If we can validate this in a larger cohort, it could redefine how we approach fusion-driven malignancies beyond Ewing sarcoma.”
For families navigating the complexities of relapsed or refractory Ewing sarcoma, access to specialized sarcoma centers with expertise in molecular diagnostics and early-phase therapeutics is critical. Institutions participating in trials like this one often provide not only cutting-edge investigational approaches but also comprehensive supportive care tailored to adolescent oncology needs. Patients and caregivers seeking expert evaluation should consider consulting with vetted board-certified pediatric oncologists who collaborate with sarcoma-focused multidisciplinary teams. Given the importance of accurate molecular profiling to confirm EWS::FLI1 status and monitor dynamic biomarkers, referral to accredited molecular pathology laboratories with validated fusion detection assays ensures eligibility for biologically informed trials. As research advances, healthcare institutions adopting adaptive trial platforms and real-time biomarker monitoring will be best positioned to deliver the next generation of precision therapies.
While these results are encouraging, they remain preliminary. The absence of a control group and the modest sample size limit definitive conclusions about efficacy. Long-term data on durability of response and late effects are still maturing. The next logical step—a randomized phase 2/3 trial comparing this regimen to standard salvage therapy or integrating it into frontline protocols for high-risk patients—is warranted. Such a study would need to incorporate patient-reported outcomes and rigorous surveillance for treatment-related sequelae to truly assess net clinical benefit.
As the field moves toward targeting transcriptional dependencies and epigenetic regulators in fusion-driven cancers, strategies like this one exemplify the promise of mechanism-driven drug repurposing and dosing innovation. Success here could catalyze similar approaches in other translocation-defined sarcomas and solid tumors where direct target inhibition remains unattainable.
Disclaimer: The information provided in this article is for educational and scientific communication purposes only and does not constitute medical advice. Always consult with a qualified healthcare provider regarding any medical condition, diagnosis, or treatment plan.