Evolutionary Drivers of Tumour Promotion
The long-held medical consensus that cancer begins with a single, catastrophic mutation is being rewritten. New evidence suggests that the seeds of malignancy are already sown in our healthy tissues, waiting for a specific environmental trigger to accelerate their growth. This shift in understanding moves the clinical focus from the act of mutation to the process of selection.
Key Clinical Takeaways:
- Normal human tissues contain millions of cells with known cancer driver mutations that typically remain dormant and maintain homeostasis.
- Many carcinogens act not as mutagens that create new mutations, but as “promoters” that modify selective constraints to favor the expansion of pre-existing mutant clones.
- Tumorigenesis is driven by Darwinian principles of variation and selection, suggesting that cancer prevention may rely on mitigating environmental promoters rather than solely avoiding mutagens.
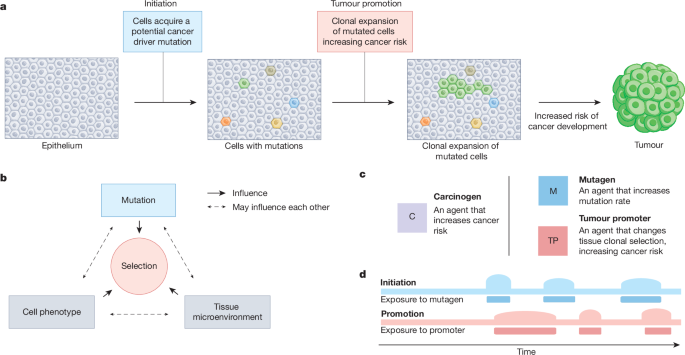
For decades, the prevailing model of carcinogenesis focused on “initiation”—the moment a mutagen damages DNA to create a driver mutation. However, this fails to explain a critical clinical paradox: why do millions of cells carrying these same driver mutations exist in healthy individuals without ever progressing to malignancy? The gap in our understanding lies in the distinction between the presence of a mutation and the selective environment that allows that mutation to thrive. Here’s the essence of tumour promotion, a process that transforms a dormant mutant cell into a clinically significant tumor.
The Initiation-Promotion Model and the Role of Selective Constraints
The conceptual foundation for this evolutionary perspective dates back to seminal research in the mid-20th century. A pivotal study by Berenblum and Shubik in 1947 demonstrated that a single application of a mutagenic carcinogen was sufficient for initiation, but it required repeated treatment with croton oil—an inflammatory agent—to actually induce skin tumors in mice. This established the initiation-promotion model, proving that although mutations are necessary, they are often insufficient on their own to cause cancer.

Modern genomics has validated this historical observation on a massive scale. We now grasp that normal human tissues are a “patchwork” of mutant clones. Most of these cells never transform because the surrounding biological environment imposes strict selective constraints that maintain homeostasis. When exogenous carcinogenic exposures occur, they often do not create new mutations; instead, they alter the cellular landscape. By modifying these constraints, promoters provide a competitive advantage to specific oncogenic clones, allowing them to outcompete healthy cells.
“The repeated interplay between variation and selection—the first principles of Darwinian evolution—underlies the clonal selection leading to tumorigenesis.”
This evolutionary trajectory is not limited to the early stages of the disease. As highlighted in research published in Cancer Discovery, cancer cells continue to adapt and survive through the acquisition and selection of molecular modifications throughout the life of the tumor. This ongoing evolution drives the progression from premalignant cells to metastatic stages and, eventually, to drug-resistant end-stage disease.
From Clonal Diversity to Malignant Progression
The pathogenesis of cancer is less like a sudden accident and more like a slow, iterative process of natural selection. In this framework, the “driver mutations” are the variation, and the environmental stressors—such as chronic inflammation or chemical exposure—are the selective pressures. When the environment shifts, cells that were previously insignificant become the most “fit” for the new conditions, leading to clonal expansion.
This realization has profound implications for how we approach risk factors. If the majority of carcinogens are not mutagens, then the traditional focus on avoiding DNA-damaging agents is only half the battle. The clinical priority must expand to include the mitigation of endogenous and environmental promoters that trigger the expansion of these pre-existing clones. For individuals with a high genetic predisposition or known exposure to promoters, early detection is critical. It is highly recommended to coordinate care with advanced molecular diagnostic centers to monitor for the expansion of premalignant clones before they reach a threshold of clinical malignancy.
The parallels between cancer and broader evolutionary biology are striking. As noted in a 2012 review in Nature, the clonal selection processes leading to malignant tumors mirror the Darwinian processes that shape biological diversity in response to genetic or environmental influences. In other words that the tumor is not a static mass of identical cells, but a diverse ecosystem of competing clones, each evolving to survive the body’s immune response and medical interventions.
Clinical Implications for Prevention and Treatment
Shifting the lens toward evolution changes the “standard of care” for cancer prevention. Rather than searching for a way to prevent every single mutation—an impossible task given the millions of driver mutations already present in healthy tissue—the goal becomes the management of the promotional environment. By eliminating the factors that grant mutant clones a competitive edge, we may be able to keep these cells in a state of permanent dormancy.

For patients managing chronic inflammatory conditions or long-term exposure to known promoters, a proactive strategy is essential. This involves a multidisciplinary approach, combining the expertise of preventative medicine specialists to minimize environmental triggers and board-certified oncologists to implement rigorous surveillance protocols. The objective is to disrupt the evolutionary momentum of the tumor before it acquires the hallmarks of malignancy, such as angiogenesis or immune evasion.
The current trajectory of research, as seen in recent publications in Trends in Cancer, is now leveraging single-cell sequencing to unravel exactly how premalignant cells transition to metastatic stages. By understanding the specific selective pressures that drive this transition, clinicians may eventually be able to “steer” cancer evolution toward a less aggressive phenotype or identify the exact moment a dormant clone begins its expansion.
The synthesis of evolutionary genetics and oncology suggests that we are moving toward a future of “evolutionary therapy.” Instead of trying to kill every cancer cell—which often selects for the most resistant clones—future treatments may focus on modifying the selective constraints of the tumor microenvironment to render the cancer cells unfit for survival. This approach promises a more sustainable method of disease management, reducing morbidity and improving long-term patient outcomes.
As we refine our understanding of clonal selection, the ability to identify and mitigate tumor promoters will become a cornerstone of public health. The transition from a mutation-centric model to an evolution-centric model provides a clearer path toward preventing the progression of cancer before it ever becomes a clinical emergency.
*Disclaimer: The information provided in this article is for educational and scientific communication purposes only and does not constitute medical advice. Always consult with a qualified healthcare provider regarding any medical condition, diagnosis, or treatment plan.*