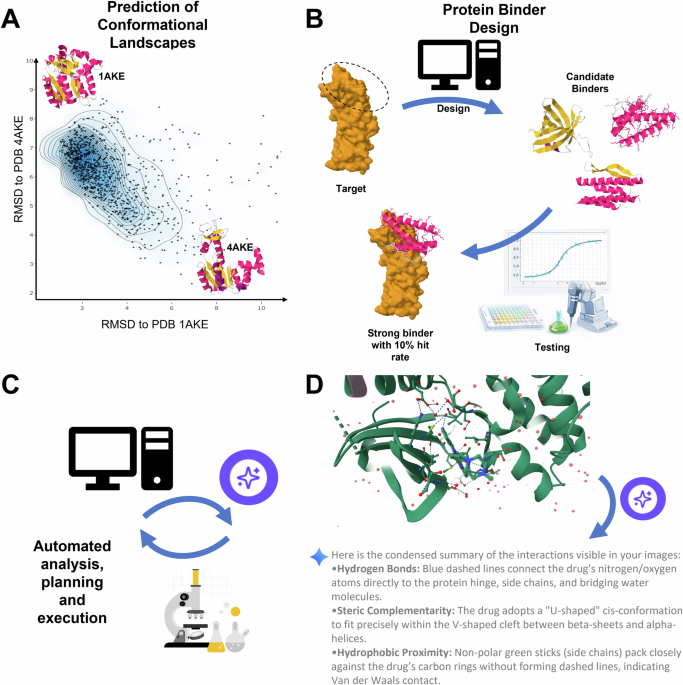
AI Breakthroughs in Protein Binder Design and Structural Biology
The structural biology community is currently attempting to replicate the “AlphaFold moment”—that specific inflection point where a seemingly intractable computational problem is solved with enough precision to render previous methodologies obsolete. While the initial breakthrough focused on the static geometry of protein domains, the current production push is targeting the dynamic: the prediction of full conformational landscapes and the systematic de novo design of high-affinity protein binders.
The Tech TL;DR:
- From Static to Dynamic: Shift from predicting a single “folded” state to mapping entire conformational landscapes using generative AI to approximate Boltzmann-weighted ensembles.
- Engineering vs. Discovery: Protein binder development is transitioning from a conceptual biological challenge into a scalable engineering discipline with higher experimental success rates.
- Computational Efficiency: New generative approaches aim to estimate free energy differences and state populations at a fraction of the compute cost required by traditional molecular dynamics.
For years, the bottleneck in structural biology has been the staggering latency of molecular dynamics (MD). Simulating the movement of a protein to understand its various states requires an immense amount of compute—often necessitating massive GPU clusters and weeks of wall-clock time to capture a few microseconds of biological activity. This is the “latency issue” of the bio-tech stack: the gap between theoretical model and experimental verification is too wide for rapid iterative development.
The Shift to Boltzmann-Weighted Ensembles
The current architectural shift involves moving away from the brute-force integration of Newton’s laws of motion. Instead, new generative models are designed to approximate Boltzmann-weighted ensembles. In technical terms, this means the AI is not simulating the path from State A to State B, but is instead directly estimating the probability distribution of all possible states.
By calculating free energy differences and state populations directly, researchers can bypass the most computationally expensive parts of the MD pipeline. This isn’t just a marginal gain in speed; it is a fundamental change in the complexity class of the problem. When you move from simulating trajectories to predicting ensembles, you reduce the requirement for raw TFLOPS and shift the burden toward model weights and inference optimization.
Implementing these pipelines at scale requires more than just a few A100s. Enterprise-grade deployment typically involves containerization via Kubernetes to manage the heavy lifting of tensor operations across distributed nodes. For firms struggling with the infrastructure overhead of these models, integrating high-performance computing (HPC) specialists is becoming a prerequisite for maintaining a competitive R&D velocity.
The Tech Stack & Alternatives Matrix
To understand where this fits in the current ecosystem, we have to compare the legacy “physics-first” approach with the emerging “AI-first” generative approach.

| Metric | Traditional Molecular Dynamics (MD) | Generative AI Ensembles | De Novo Binder Design |
|---|---|---|---|
| Compute Cost | Extremely High (Iterative) | Moderate (Inference-heavy) | Low to Moderate |
| Primary Goal | Trajectory Simulation | State Population Estimation | Binding Specificity |
| Bottleneck | Time-step integration latency | Training data quality | Experimental validation |
| Outcome | Atomic-level movement | Free energy landscapes | High-affinity binders |
Programmable Protein Design: The New Engineering Discipline
Beyond conformational landscapes, the routine de novo design of protein binders is moving into a “programmable” phase. The goal is no longer to find a protein in nature that happens to bind to a target, but to computationally architect a binder with specific binding affinities from scratch.
This transition turns biology into a software problem. If you can define the target geometry and the desired binding energy, the AI acts as the compiler, generating the amino acid sequence that fulfills those constraints. This is where the “AlphaFold moment” becomes tangible for the pharmaceutical industry: the ability to move from target identification to a high-affinity lead candidate in a fraction of the traditional time.
“The transition from discovery-based biology to engineering-based design is the single most key shift in the current biotech stack. We are moving from ‘searching for the needle’ to ‘printing the needle’.”
However, the “anti-vaporware” reality is that in silico success does not always equal in vitro success. The Critical Assessment of Structure Prediction (CASP) continues to be the gold standard for auditing these claims, ensuring that the “success rates” touted by AI platforms are backed by experimental evidence rather than just low-loss functions on a training set.
Implementation Mandate: The Design Pipeline
For developers building wrappers around these structural biology tools, the workflow typically involves a CLI-driven pipeline that moves from sequence generation to folding prediction and finally to affinity scoring. While specific proprietary APIs vary, a typical open-source inspired workflow for a binder design loop might look like this in a bash environment:

# Step 1: Generate candidate sequences based on target scaffold python design_binder.py --target target_protein.pdb --num_candidates 1000 --out ./candidates/ # Step 2: Predict 3D structures for candidates (using an AlphaFold-like inference engine) for fold in ./candidates/*.fasta; do predict_structure --input $fold --output ./folds/ --gpu 0 done # Step 3: Score binding affinity using a free-energy estimator python score_affinity.py --target target_protein.pdb --candidates ./folds/ --threshold -10.5 > high_affinity_leads.txt Scaling this process requires deep integration with CUDA-optimized libraries and efficient memory management to prevent OOM (Out-of-Memory) errors during the folding of large protein complexes. Many organizations are now outsourcing the development of these custom pipelines to specialized bioinformatics dev shops that can optimize the Python-to-C++ bridge for maximum throughput.
The Architectural Outlook
The trajectory is clear: we are moving toward a world where protein design is as iterative as frontend development. The integration of general reasoning AI agents into these platforms suggests a future where the “prompt” is a biological function, and the “output” is a validated molecular structure. The risk, as always, is the “black box” nature of these generative models. Without a grounding in the underlying physics—specifically the Boltzmann distributions mentioned in the Nature report—we risk designing proteins that look perfect on a screen but fail to fold in a wet lab.
For the CTOs and lead engineers in the biotech space, the priority is no longer just acquiring the models, but building the data flywheels and HPC infrastructure necessary to validate them. The “AlphaFold moment” was the beginning; the engineering of a programmable biological world is the actual deployment.
*Disclaimer: The technical analyses and security protocols detailed in this article are for informational purposes only. Always consult with certified IT and cybersecurity professionals before altering enterprise networks or handling sensitive data.*